Quality control
We will start by looking into quality scores of our sequences to do a quality check. To achieve this task, we use FASTQC program with raw sequencing data. FASTQC provides an easy-to-understand html report, facilitating an efficient quality check of fastq files.
The command below run the fastqc program for each fastq (or compressed fastq.gz) file and produces a report for each file in the output_directory.
fastqc fastq_directory -o output_directoryIn our case, we have multiple compressed fastq files with the file extension of .fastq.gz. We will use the expression *.fastq.gz to specify all those files for the processing. The following command run fastqc program for every file.
fastqc ./raw_data/*.fastq.gz -o ./fastqc_resultsThis execution results in the generation of a html and a zip file for each processed fastq file. The html file provides a report consisting of tables, figures about the quality check. The zip file contains all the figures, tables, and summaries. Figure fig-quality shows the distribution of quality scores for each position for a particular fastq file (SRR6915103_1).
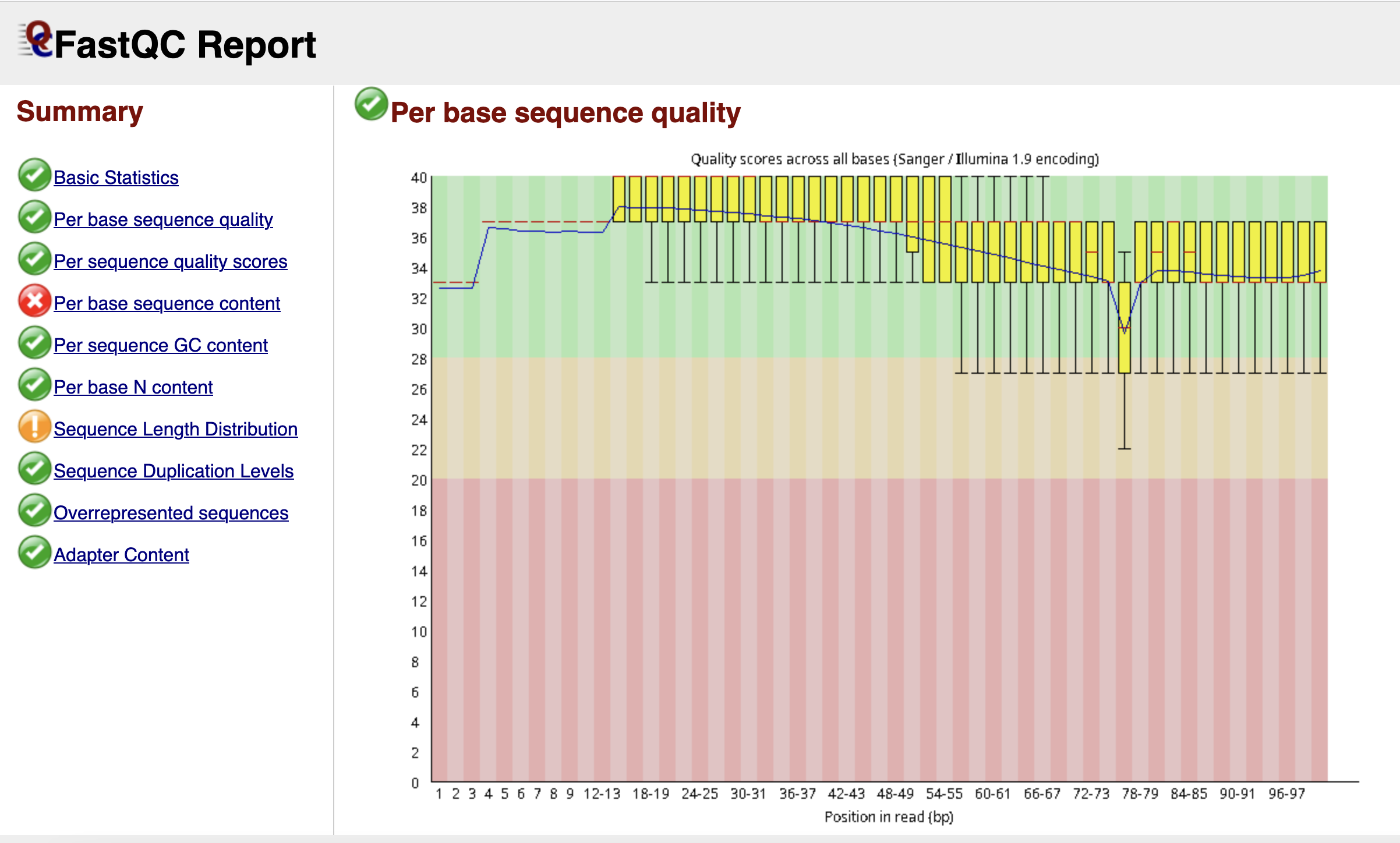
Such report is generated for each fastq file and it can become cumbersome checking each of those files for quality check. To simplify this step and aggregate all such reports into a single one, we use MULTIQC program (Ewels et al. 2016).
The command below run multiqc program which generates a html report and a directory of images, tables, and summaries.
multiqc ./fastqc_results -o multiqc_resultsThe below image shows a snapshot of multiqc report.
Trimming sequences to enhance quality
This task involves removing sequences due to low quality or due to other measures (e.g., shorter than a particular length).
We will use here trim-galore (Krueger 2015) to perform the trimming task. The program runs for each paired-eng sequence. Therefore, we will write a script to iterate over all the fastq files in the directory.
The following script provides that functionality.
#!/bin/bash
# Create output directory if it doesn't exist
mkdir -p glore_output
# Loop over all R1 files ending with _1.fastq.gz
for r1 in raw_data/*_1.fastq.gz; do
# Derive the corresponding R2 file by replacing _1 with _2
r2="raw_data/$(basename "$r1" _1.fastq.gz)_2.fastq.gz"
# Run Trim Galore for each pair and store output in glore_output
trim_galore --nextera --quality 20 --length 75 --paired "$r1" "$r2" --output_dir glore_output
done